|
The referenced media source is missing and needs to be re-embedded.
|
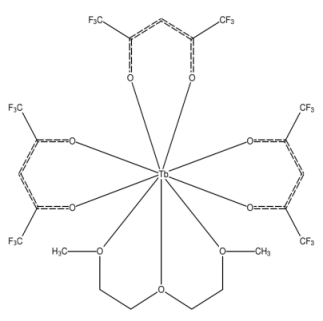
A Comprehensive Strategy to Boost the Quantum Yield of Luminescence of Europium Complexes Nathalia B. D. Lima, Simone M. C. Gonçalves, Severino A. Júnior, and Alfredo M. Simas |