Molecular magnets is a very active research area. Recently, there has been interest in the development of molecular magnets with paramagnetic lanthanide ions. For example, see the article below:
Magnetic molecular materials with paramagnetic lanthanide ions
BingWu Wang, ShangDa Jiang, XiuTeng Wang, and Song Gao
Science in China Series B: Chemistry, 2009, v.52, n.11, 1739-1758.
Double meta-substituents on pyridyl groups are known to couple ferromagnetically, leading to open shell triplet ground states. For detals, see:
Spin control in organic molecules
Dennis A. Dougherty
Acc. Chem. Res., 1991, 24 (3), pp 88–94
Thus, as an example, let us consider the complex below:

The contribution of the f-orbitals of the lanthanide to the spin density is not taken into account in this tutorial.
With the Sparkle Model, it is possible to compute the spin density of the ligands only in the presence of the lanthanide ion.
- First draw and optimize the geometry of your complex, following the instructions in Drawing Complexes.
- Replace the keyword SINGLET by the keyword TRIPLET, and add the keywords UHF and SPIN. The keyword line should now read AM1 XYZ AUX GNORM=0.25 BFGS SPARKLE CHARGE=3 Triplet UHF SPIN. Now, you must reoptimize the complex to obtain its most stable geometry in the triplet state.
- As an example, we provide the double_meta_pyridyl_eu.mop.
- Let us now visualize the spin density of the complex. After completion of the calculation, open Gabedit and click on “Display Geometry/Orbitals/Density/Vibration” icon . This will open a new window called “Gabedit: Orbitals/Density/Vibration”.
- Right-click on the black screen and choose “Orbitals” > “Read geometries and orbitals from a Mopac aux file”. Find your .aux file and click on "Open” (for convenience, we provide double_meta_pyridyl_eu.aux). If you prefer, close the "Orbitals" window.
- Right click on the black window and choose “Density” > “Spin”.
A new window “Calculation of molecular electronic spin density grid”, will open.
If you wish, you may increase the "Number of points" to improve image quality. However, a larger number of points will substantially take more CPU time to run. At first, leave the number of points at its default value of 65.
Click on "OK".
- Gabedit will start computing the spin density of the ligands. Be patient! The progress of the calculation can be checked on the bottom left part of the screen.
- The spin density will be drawn when the calculation ends:
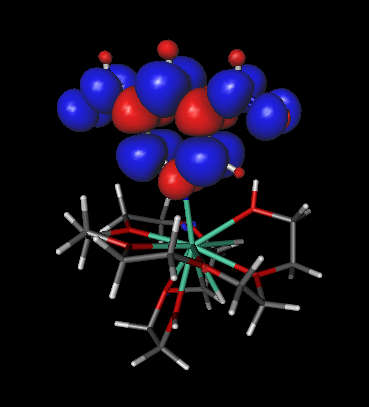
- The blue balloons indicate regions where the spin density is positive, and the red balloons the regions where the spin density is negative. In MOPAC2012, the default value of MS, when one uses the keyword TRIPLET is +1. Thus, the blue regions must outnumber the red regions. You may change the MS value using the MS keyword in MOPAC2012.